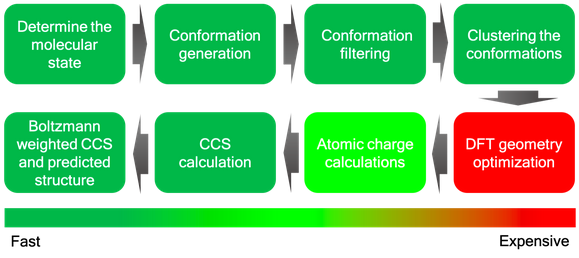
We take the neutral SMILES (Simplified Molecular Input Line Entry System) as the input to the Collisional Cross Section (CCS) prediction workflow (Figure 1). First, we model the gas phase conformational ensemble. Protonation/deprotonation isomers are hypothesized using Dimorphite-DL. [1] Protonation isomers within 10 kcal/mol are retained, determined by single point quantum mechanics energy calculation using DFT B3LYP with 6-311G(d,p) as the basis set implemented in the QUICK program. [2] Up to 1000 conformers are generated using the ETKDG algorithm implemented in RDKit. [3] The conformers are rapidly optimized using the ANI-2x deep learning potential trained on DFT calculation data. [4] From the conformers biased toward the energy minima, representative conformers are selected by performing conformational clustering through the AutoGraph algorithm. [5] The selected centroids are geometry optimized using DFT B3LYP with 6-31++G(d,p) as the basis set in QUICK. [2] The atomic charge is calculated in this step. The set of conformers unique to 0.1 Å are treated as the conformational ensemble.
The theoretical Collisional Cross Section (CCS) values are calculated for each conformer in the ensemble using the trajectory method implemented in the HPCCS program (T = 298.0 K, carrier gas = N2). [6] The ensemble average of the CCS is reported.
The performance of the workflow has been validated over 20 metabolites, exhibiting a mean relative error of 3 %. The uncertainty in experimental measurements is 3 ± 2 %, thus the performance of the workflow appears to be consistent with the limit of experimental reproducibility.
[1] Ropp, P. J.; Kaminsky, J. C.; Yablonski, S.; Durrant, J. D. J. Cheminfo. 2019, 11, 14. https://doi.org/10.1186/s13321-019-0336-9
[2] Manathunga, M.; Miao, Y.; Mu, D.; Götz, A. W.; Merz Jr., K. M. J. Chem. Theory Comput. 2020, 16, 4315. https://doi.org/10.1021/acs.jctc.0c00290
[3] Riniker, S.; Landrum, G. A. J. Chem. Inf. Model. 2015, 55, 2562. https://doi.org/10.1021/acs.jcim.5b00654
[4] Devereux, C.; Smith, J. S.; Davis, K. K.; Barros, K.; Zubatyuk, R.; Isayev, O.; Roitberg, A. E. J. Chem. Theory Comput. 2020, 16, 4192. https://doi.org/10.1021/acs.jctc.0c00121
[5] Tanemura, K. A.; Das, S.; Merz Jr. K. M. J. Chem. Inf. Model. 2021, 61, 1647. https://doi.org/10.1021/acs.jcim.0c01492
[6] Zanotto, L.; Heerdt, G.; Souza, P. C. T.; Araujo, G.; Skaf, M. S. J. Comp. Chem. 2018, 39, 1675. https://doi.org/10.1002/jcc.25199